


TRIAGEM NEONATAL (teste do pezinho)
Aula Profª Francimeiry Carvalho - UESPI, DEZEMBRO DE 2009.
CONCEITO
O termo triagem, que se origina do vocábulo francês triage, significa seleção, separação de um grupo, ou mesmo, escolha entre inúmeros elementos.
Em Saúde Pública, a ação primária dos programas de Triagem, ou seja, a detecção – através de testes aplicados numa população – de um grupo de indivíduos com probabilidade elevada de apresentarem determinadas patologias.
Rastreamento especificamente na população com idade de 0 a 30 dias de vida.
Na Triagem Neonatal, além das doenças metabólicas, podem ser incluídas outros tipos de patologias como as hematológicas, infecciosas, genéticas.
FUNDAMENTOS HISTÓRICOS
No final da década de 50, nos Estados Unidos, o biólogo Robert Guthrie passou a dirigir seus estudos para a prevenção da doença mental e, com este objetivo, adaptou o método de inibição bacteriana em que vinha trabalhando para a realização de identificação de erros inatos do metabolismo.
Através desta metodologia poder-se-ia detectar patologias que tardiamente culminavam com o retardo mental dos pacientes.
Desde a década de 60, a Organização Mundial da Saúde (OMS) preconiza a importância da realização dos programas populacionais de Triagem Neonatal, especialmente nos países em desenvolvimento, além de criar critérios para a realização dos mesmos.
No Brasil, a primeira tentativa ocorreu em 1976, na cidade de São Paulo, numa associação dedicada ao atendimento a crianças portadoras de deficiência mental (Associação de Pais e Amigos dos Excepcionais – APAE-SP)
Inicialmente realizava-se somente o diagnóstico de Fenilcetonúria, porém a partir de 1980 incorporou- se a detecção precoce do Hipotireoidismo Congênito
O Ministério da Saúde fez o lançamento, em 6 de junho de 2001 (Portaria GM/MS n.º822), do Programa Nacional de Triagem Neonatal.
O PNTN tem o objetivo de ampliar a Triagem Neonatal existente (Fenilcetonúria e Hipotireoidismo Congênito), incluindo a detecção precoce de outras doenças congênitas como as Doenças Falciformes, outras Hemoglobinopatias e a Fibrose Cística
NORMAS E RECOMENDAÇÕES PARA A ORGANIZAÇÃO E EXECUÇÃO DA COLETA DE AMOSTRAS:
RESPONSABILIDADES:
DO LABORATÓRIO ESPECIALIZADO DO SERVIÇO DE REFERÊNCIA EM TRIAGEM NEONATAL/LABORATÓRIO ESPECIALIZADO deve:
Identificar e capacitar um número de postos de coleta suficientes, de forma a permitir o acesso fácil da população em toda a sua área de responsabilidade;
Distribuir lanceta e papel filtro padronizado, de maneira a não haver solução de continuidade na rede;
Treinar os técnicos de enfermagem dos postos de coleta envolvidos com o programa;
Treinar e conscientizar os funcionários administrativos dos postos de coleta, enfocando a importância na agilidade dos procedimentos.
ATENÇÃO: EM QUALQUER CASO, A COLETA DE AMOSTRAS PARA O PROGRAMA NACIONAL DE TRIAGEM NEONATAL (PNTN) DEVE SER REALIZADA NO PERÍODO NEONATAL.
DO POSTO DE COLETA
O profissional designado como responsável pela coleta em cada Posto é a pessoa que será acionada pelo Serviço de Referência em TN toda vez que o contato com a família se fizer necessário. Geralmente é um profissional de enfermagem.
ARMAZENAGEM DO PAPEL FILTRO
O PAPEL FILTRO UTILIZADO NA TRIAGEM É DELICADO E REQUER CUIDADOS ESPECIAIS NO MANUSEIO E ARMAZENAGEM.
CALOR E UMIDADE EXCESSIVOS SÃO AS CONDIÇÕES DO AMBIENTE QUE PRECISAM SER EVITADAS, POIS PODEM SER ABSORVIDAS PELO PAPEL FILTRO, SEM QUE SE PERCEBA.
AMBIENTE DE COLETA
•Luvas de procedimento (não é necessário o uso de luvas cirúrgicas).
• Lanceta estéril descartável com ponta triangular de aproximadamente 2,0 mm.
• Recipiente (pissete) com álcool a 70% para assepsia.
• Algodão e/ou gaze pequena esterilizada.
• Papel filtro do PNTN.
COLETA - ASSEPSIA
•Realizar a assepsia do calcanhar com algodão ou gaze levemente umedecida com álcool 70%.
•Massagear bem o local, ativando a circulação.
•Certificar-se de que o calcanhar esteja avermelhado
•Aguardar a secagem completa do álcool.
*(FOTO 1)

COLETA - PUNÇÃO
•Um procedimento seguro evita complicações. A punção deve ser executada numa das laterais da região plantar do calcanhar, locais com pouca possibilidade de se atingir o osso;
*(FOTO2)

•Segure o pé e o tornozelo da criança, envolvendo com o dedo indicador e o polegar todo o calcanhar, de forma a imobilizar, mas não prender a circulação;
•Após a assepsia e secagem do álcool, penetrar num único movimento rápido toda a ponta da lanceta (porção triangular) no local escolhido, fazendo em seguida um leve movimento da mão para a direita e esquerda, para garantir um corte suficiente para o sangramento necessário;
*(FOTO3)

•Coletas de repetição ou novas punções trazem mais dor e incômodo ao bebê e à família, do que o procedimento eficiente de uma única coleta;
•Aguarde a formação de uma grande gota de sangue;
•Retire com algodão seco ou gaze esterilizada a primeira gota que se formou. Ela pode conter outros fluidos teciduais que podem interferir nos resultados dos testes;
*(FOTO4)

•Encoste o verso do papel filtro na nova gota que se forma na região demarcada para a coleta (círculos) e faça movimentos circulares com o papel, até o preenchimento de todo o círculo;
•Deixe o sangue fluir naturalmente e de maneira homogênea no papel, evitando concentração de sangue;
•Não permita que ele coagule nem no papel nem no pezinho;
•Só desencoste o papel do pezinho quando todo o círculo estiver preenchido;
*(FOTO5)

SECAGEM DA AMOSTRA
•Temperatura Ambiente – longe do sol, em ambiente de 15 a 20oC, por cerca de 3 horas.
•Isoladas – uma amostra não pode tocar outra, nem qualquer superfície.
•Posição horizontal – mantém a distribuição do sangue de forma homogênea.
SÃO PROCEDIMENTOS DE SECAGEM PORIBIDOS:
•Temperaturas altas – exposição ao sol e secagem em cima de estufas ressecam a amostra inutilizando-a;
•Ventilação forçada – ventiladores também ressecam a amostra inutilizando-a;
•Local com manipulação de líquidos ou gases químicos – podem inutilizar a amostra;
•Empilhamento de amostras – leva à mistura de sangue entre amostras diferentes;
•Contato com superfícies – algum excesso de sangue que tenha restado na amostra, não consegue se espalhar uniformemente quando em contato com superfícies.
 *(FOTO6)
*(FOTO6)
*(FOTO7)

*(FOTO8)

*(FOTO9)

*(FOTO10)

*(FOTO11)

*(FOTO12)

*(FOTO13)

*(FOTO14)
•Todos os círculos estão totalmente preenchidos;
•A amostra tem uma cor marrom-avermelhado;
•A distribuição de material é homogênea;
•A amostra não apresenta coágulos, manchas e nem hemólise;
•A amostra não está arranhada, raspada ou amassada;
•Não há sinais de contaminação;
•Todas as informações solicitadas foram preenchidas.
REQUISITOS PARA COLETA DE MATERIAL DO RN:
•Não há necessidade de jejum para a realização da coleta;
•Após 48 horas;
•Prematuridade e transfusão são fatores restritivos na triagem da Anemia falciforme e outras Hemoglobinopatias. A amostra deverá ser coletada da forma habitual para a triagem das outras doenças e nova coleta deverá ser realizada após 90 dias do nascimento;
•Coleta de amostras de gêmeos devem ser realizadas com a máxima atenção para que não haja troca na identificação das crianças nas respectivas amostras;
•Uso de medicamentos e presença de doenças não é fator restritivo para coleta de amostras.
TESTE DO PEZINHO BÁSICO:
•Fenilcetonúria e outras Aminoacidopatias
- Determinações: PKU e Cromatografia de aminoácidos
•Hipotireoidismo Congênito
- Determinações: TSH
•Anemia Falciforme e outras hemoglobinopatias
- Determinações: HbS, HbC, HbE e talassemias
FENILCETONÚRIA
•É um erro inato do metabolismo, cujo defeito metabólico (geralmente na fenilalanina hidroxilase), leva ao acúmulo de fenilalanina (FAL) no sangue e aumento da excreção urinária de ácido Fenilpirúvico e fenilalanina. Foi a primeira doença genética a ter um tratamento realizado a partir de terapêutica dietética específica;
•Para que o aumento da FAL possa ser detectado, é fundamental que a criança tenha tido ingestão protéica;
•Portanto é recomendado que a coleta seja feita após 48 horas do nascimento da criança. Nesse momento, mesmo crianças de risco, que ainda não tiveram contato com leite materno, podem colher material desde que estejam sob dieta parenteral (rica em aminoácidos essenciais).
VALOR DE REFERÊNCIA
O valor de referência da triagem para a população normal é de FAL menor ou igual a 4mg%.

*(FOTO15)
CONSEQUÊNCIAS :
•Deficiência mental irreversível;
•Convulsões, problemas de pele e cabelo;
•Problemas de urina e até invalidez permanente.
TRATAMENTO:
•Os lactentes recebem as fórmulas especiais e, a elas é adicionado leite integral modificado com a menor quantidade de FAL possível;
•Amamentação materna pode ocorrer desde que exista controle diário da FAL sangüínea;
•A introdução de outros alimentos deve ocorrer aos 4 meses de idade, contendo baixos teores de FAL, tais como vegetais e frutas, sempre com controle da quantidade diária permitida de ingesta de FAL.
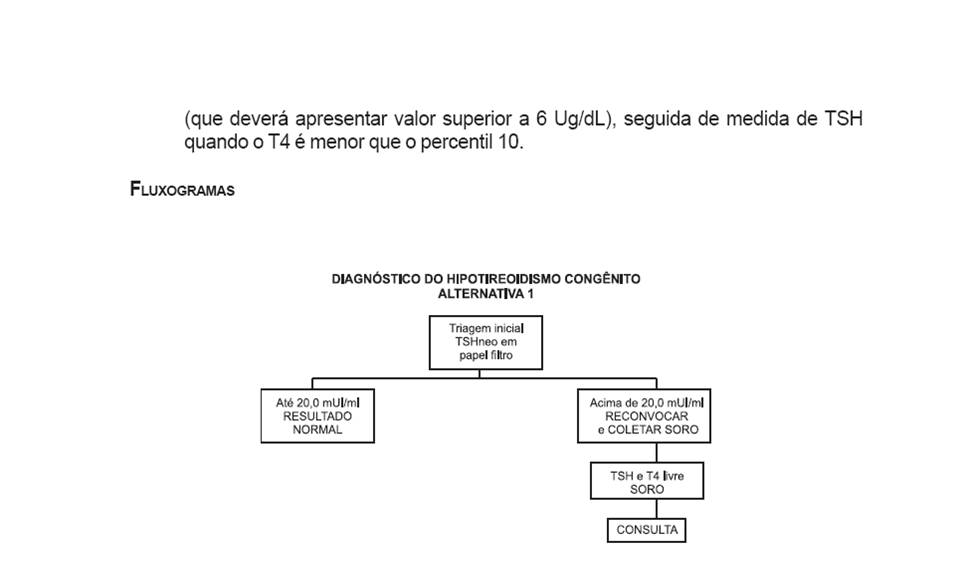
HIPOTIREOIDISMO CONGÊNITO
•De acordo com Brasil (2002), Hipotireoidismo Congênito ocorre quando a glândula tireóide do recém-nascido (RN) não é capaz de produzir quantidades adequadas de hormônios tireoidianos, o que resulta numa redução generalizada dos processos metabólicos;
•O período ideal para triagem do Hipotireoidismo Congênito é entre o quinto e sétimo dia de vida quando existe estabilização da função hormonal do recém-nascido, e possibilita diagnóstico e tratamento precoce dos casos positivos;
•Em regiões onde a deficiência de iodo não é endêmica, o Hipotiroidismo Congênito é mais freqüentemente causado pela glândula tireóide ausente ou ectópica (Hipotiroidismo Primário), de etiologia esporádica;
•Mais raramente, em cerca de 15% dos casos, é uma patologia herdada recessivamente, levando a uma falha na biossíntese do hormônio tireoidiano;
•Em crianças não submetidas a programas de Triagem Neonatal e, conseqüentemente, não tratadas precocemente, o crescimento e o desenvolvimento mental ficam seriamente comprometidos;
•As manifestações clínicas são: hipotonia muscular, dificuldades respiratórias, cianose, icterícia prolongada, constipação, bradicardia, anemia, sonolência excessiva, choro rouco, hérnia umbilical, alargamento de fontanelas, sopro cardíaco, dificuldade na alimentação com deficiente crescimento pôndero-estatural, atraso na dentição, retardo na maturação óssea, pele seca e sem elasticidade, atraso de desenvolvimento neuropsicomotor e retardo mental.
TRATAMENTO
•Consiste na reposição dos hormônios tireóideos deficitários, no caso, reposição de Levotiroxina.
•A Levotiroxina Sódica é o sal sódico do isômero sintético da Tiroxina (T4), sendo que sua utilização para reposição hormonal produz a normalização do estado metabólico que se encontra deficiente no Hipotiroidismo
*(FOTO16)
ANEMIA FALCIFORME
•É uma doença genética, devido a um defeito estrutural da cadeia Beta da globina, conduzindo a alteração físico-química na molécula de hemoglobina e na forma das hemácias para uma forma de foice, na ausência de oxigênio.
MANIFESTAÇÕES CLÍNICAS
•Anemia Hemolítica, crises de dor em membros, costas, abdomên e tórax, Insuficiência Renal Progressiva,
•Acidente vascular cerebral, maior susceptibilidade à infecções;
• Pode ocorrer alterações no desenvolvimento neurológico;
•O pico de morbidade e mortalidade está ao redor de 2 a 3 anos de vida. As principais causas de morte são septicemia e choque (por Streptococus pneumoniae ou haemophilus influenzae);
•É recomendada a detecção e início de tratamento antes de 4 meses de vida para a adequada prevenção das infecções e outras complicações que freqüentemente podem levar à morte da criança.
TESTE DO PEZINHO AMPLIADO:
•Fenilcetonúria e outras Aminoacidopatias
- Determinações: PKU e Cromatografia de aminoácidos;
•Hipotireoidismo Congênito
- Determinações: TSH e T4;
•Anemia Falciforme e outras hemoglobinopatias
- Determinações: HbS, HbC, HbE e talassemias;
•Hiperplasia Adrenal Congênita
- Determinações: 17-OH-Progesterona;
•Fibrose Cística - Determinações: IRT.
FIBROSE CÍSTICA
•É a doença hereditária severa mais comum, autossômica recessiva, que afeta especialmente pulmões e pâncreas, num processo obstrutivo por aumento da viscosidade do muco.
•Nos pulmões, o aumento da secreção bloqueia as vias aéreas propiciando a proliferação bacteriana especialmente Pseudomonas e Estafilococos), o que leva à infecção crônica, lesão pulmonar e óbito por disfunção respiratória.
SINTOMAS
•Dificuldade de ganho de peso, problemas respiratórios, perda de sal pelo suor, dor abdominal recorrente, icterícia prolongada, edema hipoproteinêmico, pancreatite recorrente, cirrose biliar.
TESTE DO PEZINHO PLUS :
•Fenilcetonúria e outras Aminoacidopatias
- Determinações: PKU e Cromatografia de aminoácidos
•Hipotireoidismo Congênito
- Determinações: TSH e T4
•Anemia Falciforme e outras hemoglobinopatias
- Determinações: HbS, HbC, HbE e talassemias
•Hiperplasia Adrenal Congênita
- Determinações: 17-OH-Progesterona
•Fibrose Cística
- Determinações: IRT
•Galactosemia
- Determinações: Galactose e Galactose-1-Fosfato
•Deficiência de Biotinidase
- Determinações: Atividade da Biotinidase
•Toxoplasmose Congênita
- Determinações: IgM anti-Toxoplasma gondii
HIPERPLASIA ADRENAL CONGÊNITA
• Distúrbio presente no nascimento, caracterizado pela deficiência nos hormônios cortisol e aldosterona e uma superprodução de andrógeno (hormônio sexual masculino).
• Os diferentes tipos de síndrome adrenogenital são hereditários, como defeitos de um gene recessivo autossômico.
• Este defeito resulta em falta de uma enzima necessária à glândula adrenal para a fabricação de cortisol.
• Em resposta a essa deficiência, a glândula pituitária secreta um hormônio (ACTH-), que estimula a glândula adrenal, causando a superprodução de hormônios andrógenos (masculinos) mas sem provocar um aumento necessário de cortisol.
• A condição afeta igualmente homens e mulheres. Em bebês do sexo feminino recém-nascidos, com este distúrbio, o clitóris se mostra dilatado com a abertura uretral na base (genitália ambígua, freqüentemente parecendo mais masculina que feminina).
• As estruturas internas do trato reprodutor (ovários, útero e tubas uterinas(Fallópio) são normais. Conforme a menina se desenvolve, a masculinização de certos traços aparece, como: voz grave, aparecimento de pêlos faciais e deficiência na menstruação na puberdade.
• Em bebês do sexo masculino recém-nascidos, nenhuma anormalidade óbvia se apresenta, mas muito antes da puberdade o menino começa a ter a massa muscular aumentada, o pênis aumenta, aparecem os pêlos púbicos e o tom da voz se torna grave.
• Os homens afetados podem parecer entrar na puberdade aos 2 ou 3 anos de idade. Na puberdade, os testículos se mostram pequenos
• O objetivo do tratamento é o retorno dos níveis hormonais de andrógeno ao normal. Isto é possível por meio da administração diária de dexametasona, fludrocortisona, ou hidrocortisona. O sexo do bebê com genitália ambígua é determinado por meio do exame dos cromossomos (cariótipo). A cirurgia reparadora em meninas é realizada entre 1 e 3 anos de idade e pode corrigir a aparência anormal.
• Os pais de crianças com esse distúrbio precisam de instrução quanto aos efeitos colaterais do tratamento com esteróides e devem ser instruídos a informar ao médico sinais de infecção e de estresse, pois pode ser necessário o aumento da dose do medicamento. Adicionalmente, o tratamento com esteróides não pode ser interrompido subitamente, por causa do risco de recorrência da insuficiência adrenal.
GALACTOSEMIA
•Incapacidade do organismo para utilizar (metabolizar) a galactose do açúcar simples (na forma de galactose 1-fosfato), que então atinge altos níveis no organismo e causa lesões a vários sistemas do organismo.
•As pessoas que sofrem de galactosemia não toleram o leite em nenhuma forma (humano e de nenhum outro tipo), já que a lactose do açúcar do leite (um dissacarídeo) é formada por partes iguais de glicose e galactose.
•A exposição ao leite gera níveis tóxicos de galactose na criança que conduz ao desenvolvimento de lesões hepáticas, formação de catarata e lesão cerebral.
•O típico quando um recém-nascido sofre de galactosemia e é alimentado com leite, é que ele desenvolva icterícia, vômitos, letargia, irritabilidade e convulsões.
• A alimentação continuada com produtos lácteos levará ao desenvolvimento de cirrose, formação de catarata e retardo mental.
TRATAMENTO:
•Uma vez diagnosticada a doença, o tratamento consiste na abstinência total de todos os tipos de leite e dos laticínios. O bebê pode ser alimentado com fórmulas baseadas em soja, carne ou Nutramigen (uma fórmula processada à base de proteína hidrolizada).
•A galactosemia é uma condição que dura a vida toda e que exige a abstinência permanente do consumo destes produtos.
DEFICIÊNCIA DA BIOTINIDASE
•É uma doença metabólica tratável na qual o organismo não consegue obter a vitamina BIOTINA da maneira adequada.
•A biotina livre é necessária para que um grupo de enzimas chamado carboxilases funcione perfeitamente.
•Carboxilases são importantes no metabolismo de algumas gorduras, carboidratos e proteínas.
•Sem a ingestão de biotina, crianças com deficiência da biotinidase podem desenvolver um ou mais dos sintomas abaixo relacionadas:
- Alopécia (queda de cabelo);
- Atraso de desenvolvimento;
- Hipotonia;
- Convulsões;
- Erupções de pele/ infecções de pele;
- Conjuntivite; - Perda de audição;
- Letargia;
-Problemas respiratórios;
- Problemas de visão;
- Coma;
- Dificuldade para alimentação;
- Infecções fúngicas;
- Hepatomegalia (aumento do fígado);
- Problemas de fala;
- Esplenomegalia (aumento do baço).
•Todas as crianças que desenvolveram os sintomas da deficiência da biotinidase tiveram significativas e rápidas melhoras com o inicio da ingestão de biotina.
TOXOPLASMOSE CONGÊNITA
•Se caracteriza pelos danos que provoca aos olhos, ao sistema nervoso, à pele e aos ouvidos.
•O recém-nascido quase sempre apresenta peso baixo ao nascer, fígado e baço dilatados, icterícia, anemia, petéquias (manchas vermelhas e delicadas na pele, provocadas pelo sangramento dos capilares) e prejuízo ocular evidenciado por inflamação da retina.
TRATAMENTO
•Na gestante, a infecção exige um tratamento agressivo, com drogas eficazes como:
- Espiramicina
- Pirimetamina
- Drogas à base de sulfa
•O tratamento de uma criança com toxoplasmose congênita é determinado com base na extensão do envolvimento da doença.
DEFICIÊNCIA DE GLICOSE 6-FOSFATO DESIDROGENASE
•Defeito hereditário das enzimas, ligado ao sexo e que resulta na ruptura dos glóbulos vermelhos quando a pessoa sofre um estresse por uma infecção ou por causa de certos medicamentos.
SINTOMAS
• Fadiga
•Cor pálida
•Falta de ar
•Freqüência cardíaca acelerada
•Coloração amarelada da pele (icterícia)
•Urina escura
•Baço aumentado
MEDICAMENTOS QUE PODEM PRECIPITAR ESAT REAÇÃO, INCLUEM:
•Agentes antimaláricos;
•Sulfonamidas (antibióticos);
•Aspirina;
•Medicamentos antiinflamatórios não esteróides (NSAIDs);
•Nitrofurantoína;
•Quinidina;
•Quinina;
•Outros;
•O distúrbio também pode resultar da: exposição a certas substâncias químicas, como aquelas presentes nas bolas de naftalina.
TESTE DO PEZINHO MASTER:
•Fenilcetonúria e outras Aminoacidopatias
-Determinações: PKU e Cromatografia de aminoácidos
•Hipotireoidismo Congênito
-Determinações: TSH e T4
•Anemia Falciforme e outras hemoglobinopatias
-Determinações: HbS, HbC, HbE e talassemias
•Hiperplasia Adrenal Congênita
-Determinações: 17-OH-Progesterona
•Galactosemia
-Determinações: Galactose e Galactose-1-Fosfato
•Deficiência de Biotinidase
-Determinações: Atividade da Biotinidase
•Toxoplasmose Congênita
-Determinações: IgM anti-Toxoplasma gondii
•Deficiência de Glicose-6-Fósforo Desidrogenase
-Determinações: Atividade da glicose-6-fosfato-desidrogenase
•Citomegalovirose Congênita
-Determinações: IgM anti-Citomegalovírus
•Doença de Chagas Congênita
-Determinações: Anticorpos totais anti-Trypanosoma cruzi
•Rubéola Congênita
-Determinações: IgM anti-vírus da Rubéola
•Fibrose Cística
-Determinações: IRT
•Sífilis Congênita
-Determinações: IgM anti-Treponema pallidum
*FOTOS : FONTE GOOGLE IMAGENS
FAZER DOWNLOAD DESTE ARQUIVO!

Nenhum comentário:
Postar um comentário
Faça críticas, elogios ou deixe sua sugestão! Seu comentário tem muito valor aqui.